IBRANCE: cápsula de palbociclib
Pfizer Laboratories Div Pfizer Inc
----------
INFORMACIÓN DE PRESCRIPCIÓN COMPLETA
1 INDICACIONES Y USO
IBRANCE es indicado para el tratamiento de pacientes adultos con cáncer de mama avanzado o metastásico con receptor hormonal (HR) positivo y receptor 2 del factor de crecimiento epidérmico humano (HER2) negativo en combinación con:
- un inhibidor de la aromatasa como tratamiento inicial a base de hormonas; o
- fulvestrant en pacientes con progresión de la enfermedad después del tratamiento hormonal.
2 POSOLOGÍA Y ADMINISTRACIÓN
2.1 Dosis y pauta terapéutica recomendadas
La dosis recomendada de IBRANCE es una cápsula de 125 mg por vía oral una vez al día por 21 días consecutivos seguidos de 7 días de descanso, lo que comprende un ciclo completo de 28 días. IBRANCE debe tomarse con alimentos [consultar Farmacología clínica (12.3)].
Administrar la dosis recomendada del inhibidor de la aromatasa que se da con IBRANCE. Consulte la Información de prescripción completa del inhibidor de la aromatasa que se usa.
Cuando se administra junto con IBRANCE, la dosis recomendada de fulvestrant es de 500 mg administrados los días 1, 15, 29 y una vez por mes posteriormente. Consulte la Información de prescripción completa del fulvestrant.
Se debe recomendar a los pacientes que tomen la dosis de IBRANCE aproximadamente a la misma hora cada día.
Si el paciente vomita o se salta una dosis, no debe tomar una dosis adicional. La siguiente dosis prescrita se deberá tomar a la hora habitual. Las cápsulas de IBRANCE deben tragarse enteras (sin masticarlas, triturarlas ni abrirlas antes de tragarlas). No se deberán ingerir las cápsulas si están rotas, agrietadas o no están intactas de alguna otra forma.
Las mujeres pre/perimenopáusicas tratadas con el tratamiento combinado de IBRANCE más un inhibidor de la aromatasa o fulvestrant se deben tratar también con agonistas de la hormona liberadora de la hormona luteinizante (LHRH) de acuerdo con las pautas de la práctica clínica actual.
En el caso de hombres tratados con la terapia combinada de IBRANCE e inhibidor de la aromatasa, considerar un tratamiento con un agonista de la LHRH de acuerdo con las pautas de la práctica clínica actual.
2.2 Modificación de la dosis
En las tablas 1, 2 y 3 se indican las modificaciones de dosis recomendadas para reacciones adversas.
75 mg/día
- *
- Si se necesita otra reducción a una dosis inferior a 75 mg/día, suspender el tratamiento.
- *
- La tabla corresponde a todas las reacciones adversas hematológicas excepto linfopenia (a menos que esté asociada a eventos clínicos, p. ej., infecciones oportunistas).
- †
- Recuento absoluto de neutrófilos (RAN): Grado 1: RAN < LIN - 1500/mm3; Grado 2: RAN 1000 - <1500/mm3; Grado 3: RAN 500 - <1000/mm3; Grado 4: RAN <500/mm3.
Controlar los recuentos completos de la sangre antes del inicio del tratamiento con IBRANCE y al comienzo de cada ciclo, así como en el día 15 de los dos primeros ciclos, y según esté indicado clínicamente.
\
Para los pacientes que presentan un grado máximo 1 o 2 de neutropenia en los primeros 6 ciclos, controlar los recuentos completos de la sangre durante los siguientes ciclos cada tres meses, antes de comenzar un ciclo y según esté indicado clínicamente.
Día 1 del ciclo:
\
No se debe administrar IBRANCE y se debe repetir el control de los recuentos completos de la sangre en el plazo de una semana. Cuando se logre la recuperación a grado ≤2, iniciar el ciclo siguiente a la misma dosis.
\
Día 15 de los dos primeros ciclos:
\
Si es el grado 3 el día 15, continuar tomando IBRANCE con la dosis actual para completar el ciclo y repetir el hemograma completo el día 22.
\
Si el grado es 4 el día 22, consultar las pautas de modificación de dosis de
a continuación.
\
Considerar la reducción de dosis en casos de una recuperación prolongada (>1 semana) de la neutropenia de grado 3 o en casos de neutropenia de grado 3 recurrente en ciclos posteriores.
En cualquier momento:
\
No se debe administrar IBRANCE hasta la recuperación a grado ≤2.
\
Reanudar a la siguiente dosis más baja.
En cualquier momento:
\
No se debe administrar IBRANCE hasta la recuperación a grado ≤2.
\
Reanudar a la siguiente dosis más baja..
No administrar hasta que los síntomas se resuelvan a:
- Grado ≤1;
- Grado ≤2 (si no se considera un riesgo de seguridad para el paciente)
Reanudar a la siguiente dosis más baja.
Grados según la versión 4.0 de los CTCAE.
\
CTCAE = Criterios comunes para la terminología de eventos adversos.
Suspender de forma permanente IBRANCE para pacientes con enfermedad pulmonar intersticial (EPI)/neumonitis grave.
Consulte en la Información de prescripción completa las pautas de ajuste de dosis para el tratamiento hormonal coadministrado en caso de toxicidad y demás información de seguridad pertinente o contraindicaciones.
Modificaciones de dosis para uso con inhibidores potentes del CYP3A
Evitar el uso concomitante de inhibidores potentes del CYP3A y considerar una medicación concomitante alternativa sin inhibición del CYP3A o con mínima inhibición. Si se debe coadministrar a los pacientes un inhibidor potente del CYP3A, reducir la dosis de IBRANCE a 75 mg una vez al día. Si se suspende el inhibidor potente, aumentar la dosis de IBRANCE (tras 3 a 5 semividas del inhibidor) a la dosis usada antes del inicio del inhibidor potente del CYP3A [consultar Interacciones farmacológicas (7.1) y Farmacología clínica (12.3)].
Modificaciones de dosis para insuficiencia hepática
No se necesita ajuste de dosis para los pacientes con insuficiencia hepática leve o moderada (clases A y B de la escala Child-Pugh). Para los pacientes con insuficiencia hepática grave (clase C de la escala Child-Pugh), la dosis recomendada de IBRANCE es 75 mg una vez al día durante 21 días consecutivos seguidos de 7 días de descanso hasta completar el ciclo de 28 días [consultar Uso en poblaciones específicas (8.6) y Farmacología clínica (12.3)].
3 FORMAS FARMACÉUTICAS Y CONCENTRACIONES
Cápsulas de 125 mg: cápsulas de gelatina dura opacas, tamaño 0, ambas partes color caramelo, con “Pfizer” impreso en tinta blanca en la parte superior y “PBC 125” en la parte inferior.
Cápsulas de 100 mg: cápsulas de gelatina dura opacas, tamaño 1, parte superior color caramelo y parte inferior color anaranjado claro, con “Pfizer” impreso en tinta blanca en la parte superior y “PBC 100” en la parte inferior.
Cápsulas de 75 mg: cápsulas de gelatina dura opacas, tamaño 2, ambas partes color anaranjado claro, con “Pfizer” impreso en tinta blanca en la parte superior y “PBC 75” en la parte inferior.
4 CONTRAINDICACIONES
Ninguna.
5 ADVERTENCIAS Y PRECAUCIONES
5.1 Neutropenia
La neutropenia fue la reacción adversa informada con más frecuencia en el Estudio 1 (PALOMA-2) con una incidencia del 80% y en el Estudio 2 (PALOMA-3) con una incidencia del 83%. Se reportaron descensos de grado ≥3 en los recuentos de neutrófilos en el 66% de los pacientes que recibían IBRANCE y letrozol en el Estudio 1 y en el 66% de los pacientes que recibían IBRANCE y fulvestrant en el Estudio 2. En el Estudio 1 y 2, la mediana de tiempo hasta el primer episodio de neutropenia de cualquier grado fue de 15 días y la mediana de duración de la neutropenia de grado ≥3 fue de 7 días [consultar Reacciones adversas (6.1)].
Controlar los recuentos completos de la sangre antes del inicio del tratamiento con IBRANCE y al comienzo de cada ciclo, así como en el día 15 de los dos primeros ciclos, y según esté indicado clínicamente. Se recomienda interrumpir la administración, reducir la dosis o retrasar el inicio de los ciclos de tratamiento para los pacientes que presenten neutropenia de grado 3 o 4 [consultar Posología y administración (2.2)].
La neutropenia febril se informó en el 1.8% de los pacientes expuestos al IBRANCE en los Estudios 1 y 2. Se observó una muerte como consecuencia de septicemia neutropénica en el Estudio 2. Los médicos deben informarles a los pacientes que notifiquen de inmediato los episodios de fiebre [consultar Información de asesoría al paciente (17)].
5.2 Enfermedad pulmonar intersticial (EPI)/Neumonitis
Los pacientes tratados con inhibidores de la cinasa dependientes de las ciclinas (CDK4/6) 4 y 6, incluido IBRANCE cuando se toma junto con tratamiento hormonal, pueden presentar enfermedad pulmonar intersticial (EPI) o neumonitis grave, potencialmente mortal o mortal.
En distintos ensayos clínicos (PALOMA-1, PALOMA-2, PALOMA-3), 1.0% de los pacientes tratados con IBRANCE presentaron EPI/neumonitis de algún grado, 0.1% tuvieron el grado 3 o 4 y no se informaron casos mortales. Se observaron casos adicionales de EPI/neumonitis en el entorno de postcomercialización, con fatalidades informadas [consultar Reacciones adversas (6.2)].
Controlar a los pacientes en caso de que presenten síntomas pulmonares de EPI/neumonitis (por ejemplo, hipoxia, tos, disnea). En el caso de los pacientes que tienen nuevos síntomas respiratorios o cuyos síntomas empeoran y se sospecha que tienen neumonitis, interrumpir IBRANCE de inmediato y evaluar al paciente. Suspender de forma permanente IBRANCE en el caso de pacientes con EPI o neumonitis graves [consultar Posología y administración (2.2)].
5.3 Toxicidad embriofetal
En función de los resultados en animales y su mecanismo de acción, IBRANCE puede causar daño fetal cuando se administra a una mujer embarazada. En estudios de reproducción en animales, la administración de palbociclib durante la organogénesis a ratas y conejas preñadas causó toxicidad embriofetal a exposiciones maternas que eran ≥4 veces la exposición clínica humana según el área bajo la curva (ABC). Advertir a las mujeres embarazadas del riesgo posible para el feto. Advertir a las mujeres con capacidad reproductiva que deben usar anticoncepción eficaz durante el tratamiento con IBRANCE y por lo menos durante 3 semanas después de la última dosis [consultar Uso en poblaciones específicas (8.1 y 8.3) y Farmacología clínica (12.1)].
6 REACCIONES ADVERSAS
Las siguientes reacciones adversas clínicamente significativas se describen en otras partes del documento:
- Neutropenia [consultar Advertencias y precauciones (5.1)]
- EPI/neumonitis [consultar Advertencias y precauciones (5.2)]
6.1 Experiencia en estudios clínicos
Como los ensayos clínicos se realizan en condiciones diversas, las tasas de reacciones adversas que se observan no se pueden comparar directamente con las tasas de otros ensayos clínicos, y podrían no reflejar las tasas que se observan en la práctica clínica.
Estudio 1: IBRANCE con letrozol
Pacientes con cáncer de mama avanzado o metastásico positivo para receptores de estrógeno (ER) y negativo para HER2, como tratamiento inicial a base de hormonas
En el Estudio 1 (PALOMA-2), se evaluó la seguridad de IBRANCE (125 mg/día) con letrozol (2.5 mg/día) frente al placebo con letrozol. Los datos que se describen a continuación reflejan la exposición a IBRANCE en 444 de 666 pacientes con cáncer de mama avanzado ER positivo, HER2 negativo que recibieron al menos una dosis de IBRANCE con letrozol en el Estudio 1. La mediana de duración del tratamiento con IBRANCE con letrozol fue de 19.8 meses, mientras que la mediana de duración del tratamiento en el grupo de placebo con letrozol fue de 13.8 meses.
Se produjeron reducciones de dosis debido a una reacción adversa de cualquier grado en el 36% de los pacientes que recibían IBRANCE con letrozol. No se permitió la reducción de la dosis de letrozol en el Estudio 1.
Se produjo suspensión permanente debido a una reacción adversa en 43 de 444 (9.7%) pacientes que recibían IBRANCE con letrozol y en 13 de 222 (5.9%) pacientes que recibían placebo con letrozol. Las reacciones adversas que provocaron la suspensión permanente para los pacientes que recibieron IBRANCE con letrozol incluyen neutropenia (1.1%) y aumento de alanina aminotransferasa (0.7%).
Las reacciones adversas más frecuentes (≥10%) de cualquier grado informadas en los pacientes del grupo que recibió IBRANCE con letrozol con una frecuencia descendente fueron neutropenia, infecciones, leucopenia, fatiga, náuseas, alopecia, estomatitis, diarrea, anemia, erupción cutánea, astenia, trombocitopenia, vómitos, disminución del apetito, piel seca, fiebre y disgeusia.
Las reacciones adversas de grado ≥3 informadas con más frecuencia (≥5%) en el caso de los pacientes que recibieron IBRANCE con letrozol con una frecuencia descendente fueron neutropenia, leucopenia, infecciones y anemia.
Las reacciones adversas (≥10%) informadas en el caso de pacientes que recibieron IBRANCE con letrozol o placebo con letrozol en el Estudio 1 se enumeran en la tabla 4.
Todos los grados
\
%
Grado 3
\
%
Grado 4
\
%
Todos los grados
\
%
Grado 3
\
%
Grado 4
\
%
Grados según la versión 4.0 de los CTCAE.
\
CTCAE = Criterios comunes para la terminología de eventos adversos; N=número de pacientes; NC=no corresponde;
- *
- Las infecciones incluyen todos los términos preferidos informados que son parte de las infecciones e infestaciones de la clasificación de órganos del sistema.
- †
- Las infecciones más frecuentes (≥1%) comprenden: nasofaringitis, infección de las vías respiratorias superiores, infección del tracto urinario, herpes orales, sinusitis, rinitis, bronquitis, gripe, neumonía, gastroenteritis, conjuntivitis, herpes zóster, faringitis, celulitis, cistitis, infección de las vías respiratorias inferiores, infección dental, gingivitis, infección cutánea, gastroenteritis vírica, infección de las vías respiratorias, infección vírica de las vías respiratorias y foliculitis.
- ‡
- La estomatitis comprende: estomatitis aftosa, queilitis, glositis, glosodinia, úlceras bucales, inflamación de la mucosa, dolor bucal, molestia bucal, dolor orofaríngeo y estomatitis.
- §
- Eventos de grado 1 : 30%; Eventos de grado 2 : 3%.
- ¶
- Eventos de grado 1 : 15%; Eventos de grado 2 : 1%.
- #
- La erupción cutánea comprende los siguientes términos preferidos: erupción cutánea, exantema maculopapuloso, exantema pruriginoso, exantema eritematoso, exantema papuloso, dermatitis, dermatitis acneiforme y erupción cutánea tóxica.
Las reacciones adversas adicionales con una incidencia general del <10.0% de los pacientes que recibieron IBRANCE con letrozol en el Estudio 1 incluyeron aumento de alanina aminotransferasa (9.9%), aumento de aspartato aminotransferasa (9.7%), epistaxis (9.2%), aumento del lagrimeo (5.6%), sequedad ocular (4.1%), visión nublada (3.6%) y neutropenia febril (2.5%).
Todos los grados
\
%
Grado 3
\
%
Grado 4
\
%
Todos los grados
\
%
Grado 3
\
%
Grado 4
\
%
Estudio 2: IBRANCE con fulvestrant
Pacientes con cáncer de mama avanzado o metastásico HR positivo y HER2 negativo que presentaron progresión de la enfermedad durante o después del tratamiento previo hormonal adyuvante o para la metástasis
En el Estudio 2 (PALOMA-3), se evaluó la seguridad de IBRANCE (125 mg/día) con fulvestrant (500 mg) frente al placebo con fulvestrant. Los datos que se describen a continuación reflejan la exposición a IBRANCE en 345 de 517 pacientes con cáncer de mama avanzado o metastásico HR positivo y HER2 negativo que recibieron al menos 1 dosis de IBRANCE con fulvestrant en el Estudio 2. La mediana de duración del tratamiento con IBRANCE con fulvestrant fue de 10.8 meses, mientras que la mediana de duración del tratamiento en el grupo de placebo con fulvestrant fue de 4.8 meses.
Se produjeron reducciones de dosis debido a una reacción adversa de cualquier grado en el 36% de los pacientes que recibían IBRANCE con fulvestrant. No se permitió la reducción de la dosis de fulvestrant en el Estudio 2.
Se produjo suspensión permanente debido a una reacción adversa en 19 de 345 (6%) pacientes que recibían IBRANCE con fulvestrant y en 6 de 172 (3%) pacientes que recibían el placebo con fulvestrant. Las reacciones adversas que llevaron a la suspensión para los pacientes que recibían IBRANCE y fulvestrant incluyeron fatiga (0.6%), infecciones (0.6%) y trombocitopenia (0.6%).
Las reacciones adversas más frecuentes (≥10%) de cualquier grado reportadas en pacientes del grupo que recibía IBRANCE con fulvestrant con frecuencia descendente fueron neutropenia, leucopenia, infecciones, fatiga, náuseas, anemia, estomatitis, diarrea, trombocitopenia, vómitos, alopecia, erupción cutánea, disminución del apetito y fiebre.
Las reacciones adversas de grado ≥3 informadas con más frecuencia (≥5%) en el caso de los pacientes que recibieron IBRANCE con fulvestrant con una frecuencia descendente fueron neutropenia y leucopenia.
Las reacciones adversas (≥10%) informadas en el caso de pacientes que recibieron IBRANCE con fulvestrant o placebo con fulvestrant en el Estudio 2 se enumeran en la tabla 6.
Grados según la versión 4.0 de los CTCAE.
\
CTCAE = Criterios comunes para la terminología de eventos adversos; N=número de pacientes; NC=no corresponde.
- *
- Las infecciones incluyen todos los términos preferidos informados que son parte de las infecciones e infestaciones de la clasificación de órganos del sistema.
- †
- Las infecciones más frecuentes (≥1%) comprenden: nasofaringitis, infección de las vías respiratorias superiores, infección del tracto urinario, bronquitis, rinitis, gripe, conjuntivitis, sinusitis, neumonía, cistitis, herpes orales, infección de las vías respiratorias, gastroenteritis, infección dental, faringitis, infección ocular, herpes común y paroniquia.
- ‡
- La estomatitis comprende: estomatitis aftosa, queilitis, glositis, glosodinia, úlceras bucales, inflamación de la mucosa, dolor bucal, molestia bucal, dolor orofaríngeo, estomatitis.
- §
- Eventos de grado 1 – 17%; Eventos de grado 2 – 1%.
- ¶
- Eventos de grado 1 – 6%.
- #
- La erupción cutánea comprende erupción cutánea, exantema maculopapuloso, exantema pruriginoso, exantema eritematoso, exantema papuloso, dermatitis, dermatitis acneiforme, erupción cutánea tóxica.
Las reacciones adversas adicionales con una incidencia general del <10.0% de los pacientes que recibieron IBRANCE con fulvestrant en el Estudio 2 incluyeron astenia (7.5%), aumento de aspartato aminotransferasa (7.5%), disgeusia (6.7%), epistaxis (6.7%), aumento del lagrimeo (6.4%), sequedad cutánea (6.1%), aumento de alanina aminotransferasa (5.8%), visión borrosa (5.8%), sequedad ocular (3.8%) y la neutropenia febril (0.9%).
Todos los grados
\
%
Grado 3
\
%
Grado 4
\
%
Todos los grados
\
%
Grado 3
\
%
Grado 4
\
%
6.2 Experiencia postcomercialización
Se identificaron las siguientes reacciones adversas durante el uso de IBRANCE después de su aprobación. Dado que estas reacciones se informaron voluntariamente en una población de tamaño desconocido, no siempre es posible estimar con precisión su frecuencia ni establecer una relación causal a la exposición del fármaco.
Trastornos respiratorios: Enfermedad pulmonar intersticial (EPI)/Neumonitis no infecciosa.
Pacientes hombres con cáncer de mama avanzado o metastásico HR positivo, HER2 negativo
Según los datos limitados de los informes de uso postcomercialización y de las historias clínicas electrónicas, el perfil de seguridad para los hombres tratados con IBRANCE es coherente con el perfil de seguridad para las mujeres tratadas con IBRANCE.
7 INTERACCIONES FARMACOLÓGICAS
El palbociclib se metaboliza principalmente por el CYP3A y la enzima sulfotransferasa (SULT) SULT2A1. In vivo, el palbociclib es un inhibidor del CYP3A dependiente del tiempo.
7.1 Agentes que pueden aumentar las concentraciones plasmáticas del palbociclib
Efecto de los inhibidores del CYP3A
La coadministración de un inhibidor potente del CYP3A (itraconazol) aumentó la exposición plasmática del palbociclib en sujetos sanos en un 87%. Evitar el uso concomitante de inhibidores potentes del CYP3A (p. ej., claritromicina, indinavir, itraconazol, ketoconazol, lopinavir/ritonavir, nefazodona, nelfinavir, posaconazol, ritonavir, saquinavir, telaprevir, telitromicina y voriconazol). Evitar consumir toronja (pomelo) o el jugo de toronja durante el tratamiento con IBRANCE. Si no se puede evitar coadministrar IBRANCE con un inhibidor potente del CYP3A, se debe reducir la dosis de IBRANCE [consultar Posología y administración (2.2) y Farmacología clínica (12.3)].
7.2 Agentes que pueden disminuir las concentraciones plasmáticas del palbociclib
Efectos de los inductores del CYP3A
La coadministración de un inductor potente del CYP3A (rifampicina) disminuyó la exposición plasmática del palbociclib en sujetos sanos en un 85%. Evitar el uso concomitante de inductores potentes del CYP3A (p. ej., fenitoína, rifampicina, carbamazepina, enzalutamida y hierba de San Juan) [consultar Farmacología clínica (12.3)].
7.3 Fármacos cuyas concentraciones plasmáticas pueden verse alteradas por el palbociclib
La coadministración de midazolam con dosis múltiples de IBRANCE aumentó la exposición plasmática del midazolam en un 61%, en sujetos sanos, comparada con la administración del midazolam solo. Es posible que haya que reducir la dosis del sustrato sensible del CYP3A con un índice terapéutico estrecho (por ejemplo, afentanilo, ciclosporina, dihidroergotamina, ergotamina, everolimus, fentanilo, pimozida, quinidina, sirolimus y tacrolimus) ya que IBRANCE puede aumentar su exposición [consultar Farmacología clínica (12.3)].
8 USO EN POBLACIONES ESPECÍFICAS
8.1 Embarazo
Resumen de riesgos
En función de los resultados de estudios en animales y su mecanismo de acción, IBRANCE puede causar daño fetal cuando se administra a una mujer embarazada [consultar Farmacología clínica (12.1)]. No se dispone de datos en mujeres embarazadas para informar sobre el riesgo asociado al fármaco. En los estudios sobre la reproducción en animales, la administración de palbociclib durante la organogénesis a ratas y conejas preñadas causó toxicidad embriofetal a exposiciones maternas que eran ≥4 veces la exposición clínica humana de acuerdo con el ABC [consultar Datos]. Advertir a las mujeres embarazadas del riesgo posible para el feto.
Se desconoce el riesgo de base estimado de defectos congénitos importantes y de aborto espontáneo para la población indicada. En la población general estadounidense, el riesgo de base estimado de defectos congénitos importantes y de aborto espontáneo de los embarazos con reconocimiento clínico es del 2 al 4% y del 15 al 20%, respectivamente.
Datos
Datos en animales
En un estudio de fertilidad y desarrollo embrionario temprano en ratas hembra, se administró palbociclib por vía oral por 15 días antes del apareamiento hasta el día 7 de gestación, lo que no produjo toxicidad embrionaria en dosis de hasta 300 mg/kg/día con exposiciones sistémicas maternas de aproximadamente 4 veces la exposición humana (ABC) a la dosis recomendada.
En estudios de desarrollo embriofetal en ratas y conejas, las hembras preñadas recibieron durante el período de organogénesis dosis orales de hasta 300 mg/kg/día y 20 mg/kg/día de palbociclib, respectivamente. La dosis tóxica para las madres de 300 mg/kg/día fue fetotóxica en ratas, produciendo menores pesos corporales fetales. Con dosis ≥100 mg/kg/día en ratas, hubo una mayor incidencia de una variación ósea (mayor incidencia de una costilla presente en la séptima vértebra cervical). A la dosis tóxica para las madres de 20 mg/kg/día en conejas, hubo una mayor incidencia de variaciones óseas, incluidas falanges pequeñas en las extremidades anteriores. A 300 mg/kg/día en ratas y 20 mg/kg/día en conejas, las exposiciones sistémicas maternas fueron de aproximadamente 4 y 9 veces la exposición humana (ABC) a la dosis recomendada, respectivamente.
Se ha reportado que los ratones con inactivación doble de la CDK4/6 mueren en las etapas finales del desarrollo fetal (día de gestación 14.5 hasta el nacimiento) debido a anemia grave. Sin embargo, los datos procedentes de estos ratones pueden no ser predictivos de los efectos en seres humanos debido a diferencias en el grado de inhibición buscado.
8.2 Lactancia
Resumen de riesgos
No existen datos sobre la presencia de palbociclib en la leche humana, ni sus efectos sobre la producción de leche o el lactante alimentado con leche materna. Debido a la posibilidad de la aparición de reacciones adversas graves en lactantes alimentados con leche materna, causadas por IBRANCE, advertir a las mujeres en período de lactancia que no deben amamantar durante el tratamiento con IBRANCE y por 3 semanas después de la última dosis.
8.3 Mujeres y hombres con capacidad reproductiva
Pruebas de embarazo
IBRANCE puede causar daño fetal cuando se administra a una mujer embarazada [consultar Uso en poblaciones específicas (8.1)]. Las mujeres con capacidad reproductiva deben hacerse una prueba de embarazo antes de comenzar el tratamiento con IBRANCE.
Anticoncepción
Mujeres
IBRANCE puede causar daño fetal cuando se administra a una mujer embarazada [consultar Uso en poblaciones específicas (8.1)]. Advertir a las mujeres con capacidad reproductiva que deben usar anticoncepción eficaz durante el tratamiento con IBRANCE y por al menos 3 semanas después de la última dosis.
Hombres
Debido a la posibilidad de genotoxicidad, advertir a los hombres con parejas de sexo femenino con capacidad reproductiva que deben usar anticoncepción eficaz durante el tratamiento con IBRANCE y por 3 meses después de la última dosis [consultar Toxicología no clínica (13.1)].
Infertilidad
Hombres
De acuerdo con los estudios realizados en animales, el tratamiento con IBRANCE puede alterar la fertilidad de los hombres con capacidad reproductiva [consultar Toxicología no clínica (13.1)].
8.4 Uso pediátrico
No se estudió la seguridad ni la eficacia de IBRANCE en pacientes pediátricos.
En el estudio de toxicología de 27 semanas con dosis repetidas en ratas, se identificó alteración del metabolismo de la glucosa (glucosuria, hiperglucemia, disminución de la insulina) asociada a cambios pancreáticos (vacuolación de las células de los islotes), oculares (cataratas, degeneración del cristalino), renales (vacuolación tubular, nefropatía progresiva crónica) y del tejido adiposo (atrofia); estos cambios fueron más prevalentes en machos con dosis ≥30 mg/kg/día (aproximadamente 11 veces la exposición de adultos humanos [ABC] a la dosis recomendada). Algunos de estos hallazgos (glucosuria/hiperglucemia, vacuolación de las células de los islotes pancreáticos y vacuolación tubular renal) estuvieron presentes en el estudio de toxicología de 15 semanas con dosis repetidas en ratas inmaduras, pero con menor incidencia y gravedad. La alteración del metabolismo de la glucosa o los cambios asociados al páncreas, los ojos, los riñones y el tejido adiposo no se identificaron en un estudio toxicológico de dosis repetidas de 27 semanas con ratas que eran maduras al comienzo del estudio y con perros en estudios toxicológicos de dosis repetidas de hasta 39 semanas.
Se observaron toxicidades dentales independientes de la alteración del metabolismo de la glucosa en ratas. La administración de 100 mg/kg de palbociclib durante 27 semanas (aproximadamente 15 veces la exposición de adultos humanos [ABC] con la dosis recomendada) tuvo como resultado anomalías en los incisivos en crecimiento (decoloración, degeneración/necrosis de los ameloblastos, infiltración celular mononuclear). No se evaluaron en animales jóvenes otras toxicidades de posible preocupación para pacientes pediátricos.
8.5 Uso geriátrico
De los 444 pacientes que recibieron IBRANCE en el Estudio 1, 181 pacientes (41%) tenían ≥65 años de edad y 48 pacientes (11%) tenían ≥75 años de edad. De los 347 pacientes que recibieron IBRANCE en el Estudio 2, 86 pacientes (25%) tenían ≥65 años de edad y 27 pacientes (8%) tenían ≥75 años de edad. No se observaron diferencias generales en la seguridad ni la eficacia de IBRANCE entre estos pacientes y pacientes más jóvenes.
8.6 Insuficiencia hepática
No se necesita ajuste de dosis en pacientes con insuficiencia hepática leve o moderada (clases A y B de la escala Child-Pugh). Para pacientes con insuficiencia hepática grave (clase C de la escala Child-Pugh), la dosis recomendada de IBRANCE es 75 mg una vez al día durante 21 días consecutivos seguidos de 7 días de descanso hasta completar el ciclo de 28 días [consultar Posología y administración (2.2)]. Según un ensayo farmacocinético de sujetos con diferentes grados de función hepática, la exposición no asociada al palbociclib (ABCINF no asociada) disminuyó en un 17% en sujetos con insuficiencia hepática leve (clase A de la escala Child-Pugh) y aumentó en un 34% y 77% en sujetos con insuficiencia hepática moderada (clase B de la escala Child-Pugh) y grave (clase C de la escala Child-Pugh), respectivamente, en relación con sujetos con función hepática normal. La exposición máxima del palbociclib no asociada (Cmax no asociada) aumentó en un 7%, 38% y 72% en casos de insuficiencia hepática leve, moderada y grave, respectivamente, en relación con sujetos con función hepática normal [consultar Farmacología clínica (12.3)].
Repasar la Información de prescripción completa sobre el inhibidor de aromatasa o fulvestrant para conocer las modificaciones de dosis relacionadas con la insuficiencia hepática.
8.7 Insuficiencia renal
No se necesita ajuste de dosis en pacientes con insuficiencia renal leve, moderada o grave (CrCl >15 mL/min). Según un ensayo farmacocinético de sujetos con diferentes grados de función renal, la exposición total de palbociclib (ABCINF) ) aumentó en un 39%, 42% y 31% con insuficiencia renal leve (60 mL/min ≤ CrCl <90 mL/min), moderada (30 mL/min ≤ CrCl <60 mL/min), y grave (CrCl <30 mL/min) respectivamente, en relación con sujetos con función renal normal. La exposición máxima a palbociclib (Cmax) aumentó en un 17%, 12% y 15% para la insuficiencia renal leve, moderada y grave, respectivamente, en relación con sujetos con función renal normal. No se estudió la farmacocinética de palbociclib en pacientes que necesitan hemodiálisis [consultar Farmacología clínica (12.3)].
10 SOBREDOSIS
No existe un antídoto conocido para IBRANCE. El tratamiento de la sobredosis de IBRANCE debe consistir en medidas de apoyo generales.
11 DESCRIPCIÓN
IBRANCE en cápsulas para administración oral contiene 125 mg, 100 mg o 75 mg de palbociclib, un inhibidor de la cinasa. La fórmula molecular del palbociclib es C24H29N7O2. El peso molecular es de 447.54 daltons. El nombre químico es 6-acetil-8-ciclopentil-5-metil-2-{[5-(piperazina-1-yl)piridina-2-yl]amino}pirido[2,3-d]pirimidina-7(8H)-uno, y su fórmula estructural es:

El palbociclib es un polvo de color amarillo a naranja con un pKa de 7.4 (el nitrógeno secundario de la piperazina) y 3.9 (el nitrógeno de la piridina). A un pH 4 o menor, el palbociclib se comporta como un compuesto con alta solubilidad. Por encima de un pH 4, la solubilidad de la sustancia del fármaco disminuye de forma significativa.
Excipientes: Celulosa microcristalina, lactosa monohidrato, glicolato de almidón sódico, dióxido de silicio coloidal, estearato de magnesio y cubierta de gelatina dura de las cápsulas. Las cubiertas de las cápsulas opacas de color anaranjado claro, anaranjado claro/caramelo y caramelo contienen: gelatina, óxido férrico rojo, óxido férrico amarillo y dióxido de titanio; y la tinta para impresión contiene: goma laca, dióxido de titanio, hidróxido de amonio, propilenglicol y simeticona.
12 FARMACOLOGÍA CLÍNICA
12.1 Mecanismo de acción
El palbociclib es un inhibidor de la cinasa dependiente de las ciclinas (CDK) 4 y 6. Las ciclinas D1 y CDK4/6 se encuentran en una fase posterior de las vías de señalización que conducen a la proliferación celular. In vitro, el palbociclib redujo la proliferación celular de las líneas celulares de cáncer de mama positivo para receptores de estrógeno (ER) bloqueando la progresión de la célula desde la fase G1 a la S del ciclo celular. El tratamiento de las líneas celulares de cáncer de mama con la combinación de palbociclib y antiestrógenos conduce a una menor fosforilación de la proteína de retinoblastoma (Rb), lo que produce una menor expresión de E2F y señalización, y una mayor interrupción del crecimiento en comparación con el tratamiento con cada fármaco por sí solo. El tratamiento in vitro de las líneas celulares de cáncer de mama ER positivo con la combinación de palbociclib y antiestrógenos genera una mayor senescencia celular en comparación con cada fármaco solo, que se mantuvo hasta por 6 días tras el cese del fármaco y fue mayor si el tratamiento con antiestrógeno continuaba. Los estudios in vivo usando un modelo xenográfico de cáncer de mama ER positivo derivado de un paciente demostraron que la combinación de palbociclib y letrozol aumentó la inhibición de fosforilación de la Rb, la señalización de fase posterior y el crecimiento tumoral en comparación con cada fármaco solo.
Las células mononucleares de la médula ósea humana tratadas con palbociclib en presencia o ausencia de un antiestrógeno in vitro no se volvieron senescentes y retomaron la proliferación tras la suspensión del palbociclib.
12.2 Farmacodinámica
Electrofisiología cardíaca
Se evaluó el efecto del palbociclib en el intervalo QT corregido para el ritmo cardíaco (QTc) usando electrocardiogramas (ECG) con tiempo ajustado para evaluar el cambio de los valores iniciales y los datos farmacocinéticos correspondientes en 77 pacientes con cáncer de mama. El palbociclib no tiene un gran efecto en el QTc (es decir, >20 ms) con 125 mg una vez al día durante 21 días consecutivos seguidos de 7 días de descanso hasta completar el ciclo de 28 días.
12.3 Farmacocinética
Se caracterizó la farmacocinética (FC) del palbociclib en pacientes con tumores sólidos, como el cáncer de mama avanzado, y en sujetos sanos.
Absorción
La concentración media máxima observada (Cmax) de palbociclib generalmente se observa entre las 6 y las 12 horas (tiempo en llegar a la concentración máxima, Tmax) luego de la administración oral. La biodisponibilidad media absoluta de IBRANCE tras una dosis oral de 125 mg es del 46%. En el intervalo posológico de 25 mg a 225 mg, el ABC y la Cmax aumentaron de forma proporcional con la dosis, en general. Se alcanzó el estado estacionario en el plazo de 8 días tras la administración repetida una vez al día. Con la administración repetida una vez al día, el palbociclib se acumuló con una mediana de cociente de acumulación de 2.4 (intervalo: 1.5 a 4.2).
Efecto de los alimentos: La absorción y exposición del palbociclib fueron muy bajas en ayunas en aproximadamente el 13% de la población. El consumo de alimentos aumentó la exposición del palbociclib en este pequeño subconjunto de la población, pero no alteró la exposición del palbociclib en el resto de la población en un grado que fuera de interés clínico. Por lo tanto, el consumo de alimentos redujo la variabilidad interindividual de la exposición del palbociclib, lo que respalda la administración de IBRANCE con alimentos. En comparación con IBRANCE administrado después de ayunar toda la noche, la curva del tiempo de concentración de cero a infinito (ABCINF) y la Cmax promedio poblacional del palbociclib aumentaron en un 21% y un 38%, respectivamente, cuando se administró con alimentos ricos en grasas y calorías (aproximadamente 800 a 1000 calorías con 150, 250 y 500 a 600 calorías provenientes de proteínas, hidratos de carbono y grasas, respectivamente), en un 12% y 27%, respectivamente, cuando se administró con alimentos bajos en grasas y calorías (aproximadamente 400 a 500 calorías con 120, 250 y 28 a 35 calorías provenientes de proteínas, hidratos de carbono y grasas, respectivamente), y en un 13% y 24%, respectivamente, cuando se proporcionaron alimentos con grasas moderadas y calorías estándar (aproximadamente 500 a 700 calorías con 75 a 105, 250 a 350 y 175 a 245 calorías provenientes de proteínas, hidratos de carbono y grasas, respectivamente) una hora antes y dos horas después de la administración de IBRANCE.
Distribución
La unión in vitro del palbociclib a las proteínas plasmáticas humanas fue de aproximadamente el 85%, sin que dependiera de la concentración en el intervalo de concentraciones de 500 ng/ml a 5000 ng/ml. La fracción media sin consolidar (fu) de palbociclib en plasma humano in vivo aumentó gradualmente al empeorar la función hepática. No hubo una tendencia evidente en la media de la fu en el plasma humano in vivo en caso de empeoramiento de la función renal. Este volumen aparente de la media geométrica (Vz/F) fue 2583 l con un coeficiente de variación (CV) del 26%.
Metabolismo
Los estudios in vitro e in vivo indicaron que el palbociclib se metaboliza en el hígado en los seres humanos. Tras la administración oral de una dosis única de 125 mg de [14C]palbociclib a seres humanos, las vías metabólicas principales para el palbociclib comprendían la oxidación y la sulfonación, siendo la acetilación y la glucuronidación vías secundarias. El palbociclib fue la entidad circulante principal proveniente del fármaco en el plasma (23%). El principal metabolito circulante fue un conjugado glucurónido del palbociclib, aunque solo representó el 1.5% de la dosis administrada en las excreciones. El palbociclib se metabolizó de forma extensa y el fármaco inalterado representó el 2.3% y el 6.9% de radioactividad en las heces y la orina, respectivamente. En las heces, el conjugado de ácido sulfámico del palbociclib fue el componente farmacológico principal, responsable del 26% de la dosis administrada. En estudios in vitro con hepatocitos humanos, las fracciones citosólicas y S9 hepáticas, y las enzimas SULT recombinantes indicaron que el CYP3A y la SULT2A1 están involucrados principalmente en el metabolismo del palbociclib.
Eliminación
La media geométrica de la eliminación oral aparente (CL/F) del palbociclib fue de 63.1 l/h (CV del 29%) y la semivida media (± desviación estándar) de eliminación plasmática fue de 29 (±5) horas en pacientes con cáncer de mama avanzado. En 6 hombres sanos que recibieron una dosis oral única de [14C]palbociclib, en 15 días se recuperó una mediana del 91.6% de la dosis radiactiva total administrada; las heces (74.1% de la dosis) fueron la vía principal de excreción, y el 17.5% de la dosis se recuperó en la orina. La mayoría de la sustancia se excretó en forma de metabolitos.
Edad, sexo y peso corporal
Según un análisis farmacocinético poblacional en 183 pacientes con cáncer (50 hombres y 133 mujeres, con edades comprendidas entre 22 y 89 años, y pesos corporales de 37.9 a 123 kg), el sexo no tuvo efecto sobre la exposición del palbociclib, y la edad y el peso corporal no tuvieron efecto de importancia clínica sobre la exposición del palbociclib.
Población pediátrica
No se evaluó la farmacocinética de IBRANCE en pacientes menores de 18 años.
Insuficiencia hepática
Los datos provenientes de un ensayo farmacocinético de sujetos con diferentes grados de insuficiencia hepática indicaron que la ABCINF no asociada al palbociclib disminuyó en un 17% en sujetos con insuficiencia hepática leve (clase A de la escala Child-Pugh) y aumentó en un 34% y 77% en sujetos con insuficiencia hepática moderada (clase B de la escala Child-Pugh) y grave (clase C de la escala Child-Pugh), respectivamente, en relación con sujetos con función hepática normal. La Cmax del palbociclib no asociada aumentó en un 7%, 38% y 72% en casos de insuficiencia hepática leve, moderada y grave, respectivamente, en relación con sujetos con función hepática normal. Además, según un análisis farmacocinético poblacional de 138 pacientes, en los que 40 pacientes tenían insuficiencia hepática leve en función de la clasificación del Instituto Nacional del Cáncer (NCI) estadounidense (bilirrubina total ≤ ULN y AST > ULN, o bilirrubina total >1.0 to 1.5 × ULN y cualquier AST), la insuficiencia hepática leve no tuvo ningún efecto en la exposición del palbociclib, lo cual respalda los resultados del estudio de insuficiencia hepática específico.
Insuficiencia renal
Los datos provenientes de un ensayo farmacocinético de sujetos con diferentes grados de insuficiencia renal, la ABCINF del palbociclib aumentó en un 39%, 42% y 31% con insuficiencia renal leve (60 mL/min ≤ CrCl < 90 mL/min), moderada (30 mL/min ≤ CrCl <60 mL/min), y grave (CrCl <30 mL/min) respectivamente, en relación con sujetos con función renal normal. La exposición máxima a palbociclib (Cmax) aumentó en un 17%, 12% y 15% para la insuficiencia renal leve, moderada y grave, respectivamente, en relación con sujetos con función renal normal. Además, según un análisis farmacocinético poblacional que tuvo 183 pacientes, de los cuales 73 tenían insuficiencia renal leve y 29 tenían insuficiencia renal moderada, la insuficiencia renal leve y moderada no tuvo ningún efecto en la exposición al palbociclib. No se estudió la farmacocinética del palbociclib en pacientes que necesitan hemodiálisis.
Interacciones farmacológicas
Los datos in vitro indican que el CYP3A y la enzima SULT SULT2A1 participan principalmente en el metabolismo del palbociclib. El palbociclib es un inhibidor débil dependiente del tiempo del CYP3A tras la administración diaria de 125 mg hasta el estado estacionario en seres humanos. In vitro, el palbociclib no es un inhibidor del CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19 y 2D6, y no es un inductor del CYP1A2, 2B6, 2C8 y 3A4 en concentraciones de interés clínico.
Inhibidores del CYP3A: Los datos provenientes de un ensayo clínico de interacción farmacológica en sujetos sanos (N = 12) indican que la coadministración de múltiples dosis diarias de 200 mg de itraconazol con una dosis única de 125 mg de IBRANCE aumentó el ABCINF y la Cmax del palbociclib en aproximadamente un 87% y 34%, respectivamente, en relación con una dosis única de 125 mg de IBRANCE administrada sola [consultar Interacciones farmacológicas (7.1)].
Inductores del CYP3A: Los datos provenientes de un ensayo clínico de interacción farmacológica en sujetos sanos (N = 15) indican que la coadministración de múltiples dosis diarias de 600 mg de rifampicina, un inductor potente del CYP3A, con una dosis única de 125 mg de IBRANCE disminuyó la ABCINF y la Cmax del palbociclib en un 85% y 70%, respectivamente, en relación con una dosis única de 125 mg de IBRANCE administrada sola. Los datos provenientes de un ensayo clínico de interacción farmacológica en sujetos sanos (N = 14) indican que la coadministración de múltiples dosis diarias de 400 mg de modafinilo, un inductor moderado del CYP3A, con una dosis única de 125 mg de IBRANCE disminuyó la ABCINF y la Cmax del palbociclib en un 32% y 11%, respectivamente, en relación con una dosis única de 125 mg de IBRANCE administrada sola [consultar Interacciones farmacológicas (7.2)].
Sustratos del CYP3A: El palbociclib es un inhibidor débil dependiente del tiempo del CYP3A tras la administración diaria de 125 mg hasta el estado estacionario en seres humanos. En un ensayo clínico de interacción farmacológica con sujetos sanos (N = 26), la coadministración de midazolam con dosis múltiples de IBRANCE aumentó los valores de ABCINF y Cmax del midazolam en un 61% y 37%, respectivamente, en comparación con la administración del midazolam solo [consultar Interacciones farmacológicas (7.3)].
Medicamentos que elevan el pH gástrico: En un ensayo clínico de interacción farmacológica con sujetos sanos, la coadministración de una dosis única de 125 mg de IBRANCE con dosis múltiples del inhibidor de la bomba de protones (IBP) rabeprazol con alimentos redujo la Cmax del palbociclib en un 41%, pero tuvo un impacto limitado sobre el valor de ABCINF (reducción del 13%), en comparación con una dosis única de IBRANCE administrado solo. Dado el efecto reducido sobre el pH gástrico de los antagonistas de los receptores H2 y antiácidos locales en comparación con los IBP, se espera que el efecto de estas clases de reductores de ácido sobre la exposición del palbociclib con alimentos sea mínimo. Con alimentos, no hay un efecto de interés clínico de los IBP, los antagonistas de los receptores H2 o los antiácidos locales sobre la exposición del palbociclib. En otro estudio con sujetos sanos, la coadministración de una dosis única de IBRANCE con dosis múltiples del IBP rabeprazol en ayunas disminuyó el valor de ABCINF y la Cmax del palbociclib en un 62% y un 80%, respectivamente, en comparación con una dosis única de IBRANCE administrada sola.
Letrozol: Los datos provenientes de un ensayo clínico en pacientes con cáncer de mama mostraron que no existía interacción farmacológica entre el palbociclib y el letrozol cuando los dos fármacos se administraban juntos.
Fulvestrant: Los datos provenientes de un ensayo clínico en pacientes con cáncer de mama mostraron que no existía interacción farmacológica de interés clínico entre el palbociclib y el fulvestrant cuando los dos fármacos se administraban juntos.
Goserelina: Los datos provenientes de un ensayo clínico en pacientes con cáncer de mama mostraron que no existía interacción farmacológica de interés clínico entre el palbociclib y la goserelina cuando los dos fármacos se administraban juntos.
Anastrozol o exemestano: No se dispone de datos clínicos para evaluar las interacciones farmacológicas entre el anastrozol o el exemestano y el palbociclib. No se prevé una interacción farmacológica clínicamente significativa entre el anastrozol o el exemestano y el palbociclib según los análisis de los efectos del anastrozol, el exemestano y el palbociclib en o mediante procesos metabólicos o sistemas de transporte.
Efecto del palbociclib sobre los transportadores: Las evaluaciones in vitro indicaron que el palbociclib tiene un bajo potencial de inhibición de las actividades del transportador de aniones orgánicos (OAT)1, OAT3, del transportador de cationes orgánicos (OCT)2 y de los péptidos transportadores de aniones orgánicos (OATP)1B1 de transportadores farmacológicos, OATP1B3 en concentraciones clínicamente relevantes. In vitro, el palbociclib tiene el potencial de inhibir el OCT1 en concentraciones clínicamente relevantes, así como de inhibir la glucoproteína-P (P-gp) o la proteína de resistencia del cáncer de mama (BCRP) en el tracto gastrointestinal con la dosis propuesta.
Efecto de transportadores en el palbociclib: Según los datos in vitro, el transporte mediado por la P-gp y la BCRP no es probable que afecte el grado de absorción oral del palbociclib con dosis terapéuticas.
13 TOXICOLOGÍA NO CLÍNICA
13.1 Carcinogénesis, mutagénesis, deterioro de la fertilidad
Se evaluó la carcinogénesis del palbociclib en un estudio de ratones transgénicos de 6 meses y en un estudio de ratas de 2 años. La administración oral del palbociclib durante 2 años tuvo como resultado un aumento de la incidencia de tumores celulares microgliales en el sistema nervioso central de ratas macho a una dosis de 30 mg/kg/día (aproximadamente 8 veces la exposición clínica humana basada en ABC). No se produjeron resultados neoplásicos en ratas hembra con dosis de hasta 200 mg/kg/día (aproximadamente 5 veces la exposición clínica humana basada en ABC). La administración oral de palbociclib en ratones transgénicos rasH2 machos y hembras durante 6 meses no tuvo como resultado una mayor incidencia de neoplasmas con dosis de hasta 60 mg/kg/día.
El palbociclib fue aneugénico en células de ovario de hámster chino in vitro y en la médula ósea de ratas macho que recibieron dosis ≥100 mg/kg/día durante 3 semanas. El palbociclib no fue mutágeno en un ensayo in vitro de mutación bacteriana inversa (Ames) ni fue clastógeno en el ensayo in vitro de aberraciones cromosómicas en linfocitos humanos.
En un estudio de fertilidad con ratas hembra, el palbociclib no afectó el apareamiento ni la fertilidad con ninguna dosis hasta 300 mg/kg/día (aproximadamente 4 veces la exposición clínica en seres humanos según el ABC) ni se observaron efectos adversos en los tejidos reproductivos femeninos en estudios de toxicidad con dosis repetidas de hasta 300 mg/kg/día en ratas y 3 mg/kg/día en perros (aproximadamente 6 veces y similar a la exposición humana [ABC], a la dosis recomendada, respectivamente).
En los estudios de toxicología con dosis repetidas en ratas y perros y en un estudio de fertilidad con ratas macho, se observaron los efectos adversos del palbociclib sobre la función reproductora y la fertilidad en machos. En estudios toxicológicos de dosis repetidas, los resultados asociados al palbociclib en los testículos, el epidídimo, la próstata y la vesícula seminal con ≥30 mg/kg/día en ratas y ≥0.2 mg/kg/día en perros incluyeron disminución del peso de los órganos, atrofia o degeneración, hipospermia, detritus celulares intratubulares y disminución de la secreción. Se observó la reversibilidad parcial de los efectos sobre los órganos genitales masculinos en ratas y perros después de un período sin administración de 4 y 12 semanas, respectivamente. Estas dosis en ratas y perros tuvieron como resultado aproximadamente ≥10 y 0.1 veces, respectivamente, la exposición [ABC] en humanos con la dosis recomendada. En el estudio de fertilidad y desarrollo embrionario temprano en ratas macho, el palbociclib no causó efectos sobre el apareamiento, pero se tradujo en una leve disminución de la fertilidad en relación con una motilidad y densidad espermáticas menores con 100 mg/kg/día con niveles de exposición proyectados [ABC] correspondientes a 20 veces la exposición en seres humanos a la dosis recomendada.
14 ESTUDIOS CLÍNICOS
Estudio 1: IBRANCE con letrozol
Pacientes con cáncer de mama avanzado o metastásico ER positivo y HER2 negativo, como tratamiento inicial a base de hormonas
El Estudio 1 (PALOMA-2) fue un estudio internacional multicéntrico, aleatorizado de doble ciego de grupos paralelos de IBRANCE con letrozol frente a placebo con letrozol, realizado con mujeres posmenopáusicas con cáncer de mama avanzado ER positivo y HER2 negativo, que no habían recibido un tratamiento sistémico previo para la enfermedad avanzada. Se aleatorizaron un total de 666 pacientes, en una relación 2:1, para recibir IBRANCE con letrozol o placebo con letrozol. La aleatorización se estratificó por lugar de la enfermedad (visceral frente a no visceral), intervalo sin enfermedad (metastásica "de novo" frente a ≤12 meses desde el final del tratamiento adyuvante hasta la reaparición de la enfermedad frente a >12 meses desde el final del tratamiento adyuvante hasta la reaparición de la enfermedad), y la naturaleza de los tratamientos anticancerígenos (neo)adyuvantes previos (tratamientos hormonales previos frente a sin tratamiento hormonal previo). Se administró IBRANCE en dosis de 125 mg por vía oral una vez al día por 21 días consecutivos seguidos de 7 días de descanso. Los pacientes recibieron el tratamiento del estudio hasta la progresión objetiva de la enfermedad, empeoramiento sintomático, toxicidad inaceptable, muerte o retirada del consentimiento, lo que ocurriera primero. El resultado principal de la eficacia del estudio fue la sobrevida libre de progresión (SLP) evaluada por investigadores examinada según los criterios de evaluación de respuesta en tumores sólidos, versión 1.1 (RECIST).
Los pacientes inscritos en este estudio tenían una mediana de edad de 62 años (intervalo 28 a 89). La mayoría de los pacientes (78%) eran de raza blanca y la mayoría (98%) tenía un estado funcional (EF) de ECOG (Eastern Cooperative Oncology Group) de 0 o 1. El cuarenta y ocho por ciento de los pacientes habían recibido quimioterapia y el 56% habían recibido tratamiento antihormonal neoadyuvante o adyuvante antes de su diagnóstico de cáncer de mama avanzado. El treinta y siete por ciento de los pacientes no había recibido tratamiento sistémico previo en el contexto neoadyuvante o adyuvante. La mayoría de los pacientes (97%) presentaba metástasis. El veintitrés por ciento de los pacientes solo tenía enfermedad ósea, y el 49% tenía enfermedad visceral.
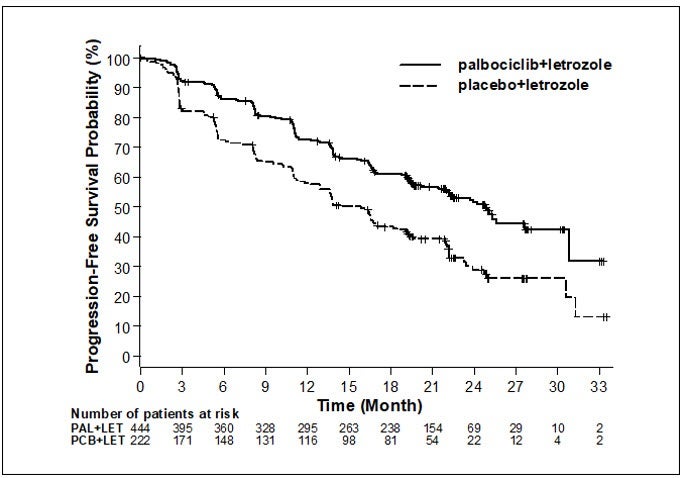
Los resultados de eficacia principales derivados del Estudio 1 se resumen en la tabla 8 y en la figura 1. Se observaron resultados consistentes entre los subgrupos de pacientes de intervalo sin enfermedad (ISE), lugar de la enfermedad y tratamiento previo. El efecto del tratamiento de la combinación en la SLP también fue respaldado por una revisión independiente de radiografías. Los datos de sobrevida general (SG) no eran consolidados en el momento del análisis final de SLP (el 20% de los pacientes habían muerto). Se continuará el seguimiento de los pacientes para el análisis final.
- *
- Respuesta basada en respuestas confirmadas.
Estudio 2: IBRANCE con fulvestrant
Pacientes con cáncer de mama avanzado o metastásico HR positivo y HER2 negativo que presentaron progresión de la enfermedad durante o después del tratamiento previo hormonal adyuvante o para la metástasis
El Estudio 2 (PALOMA-3) fue un estudio multicéntrico internacional, aleatorizado, con enmascaramiento doble, con grupos paralelos de IBRANCE con fulvestrant frente al placebo con fulvestrant en mujeres con cáncer de mama avanzado HR positivo y HER2 negativo, independientemente de su situación respecto a la menopausia, cuya enfermedad progresó durante o después del tratamiento hormonal previo. Se aleatorizaron un total 521 mujeres pre/posmenopáusicas, en una relación 2:1, para recibir IBRANCE con fulvestrant o placebo con fulvestrant y se estratificaron por sensibilidad documentada al tratamiento hormonal previo, situación respecto a la menopausia al ingresar al estudio (pre/peri frente a posmenopáusica) y presencia de metástasis en las vísceras. Se administró IBRANCE en dosis de 125 mg por vía oral una vez al día por 21 días consecutivos seguidos de 7 días de descanso. Las mujeres pre/perimenopáusicas se incorporaron al estudio y recibieron el agonista de la LHRH goserelina durante al menos 4 semanas antes y por el tiempo que duró el Estudio 2. Los pacientes continuaron recibiendo el tratamiento asignado hasta la progresión objetiva de la enfermedad, empeoramiento sintomático, toxicidad inaceptable, muerte o retirada del consentimiento, lo que ocurriera primero. El criterio principal de valoración de la eficacia del estudio fue la SLP evaluada por el investigador según la versión 1.1 de RECIST.
Los pacientes inscritos en este estudio tenían una mediana de edad de 57 años (intervalo 29 a 88). La mayoría de los pacientes del estudio eran de raza blanca (74%) y todos tenían un estado funcional (EF) de 0 o 1 según el ECOG, y el 80% eran posmenopáusicas. Todos los pacientes recibieron un tratamiento sistémico previo, y el 75% de los pacientes habían recibido un régimen de quimioterapia previo. El veinticinco por ciento de los pacientes no había recibido tratamiento previo en el contexto de enfermedad metastásica, el 60% tenía metástasis visceral y el 23% tenía enfermedad ósea únicamente.
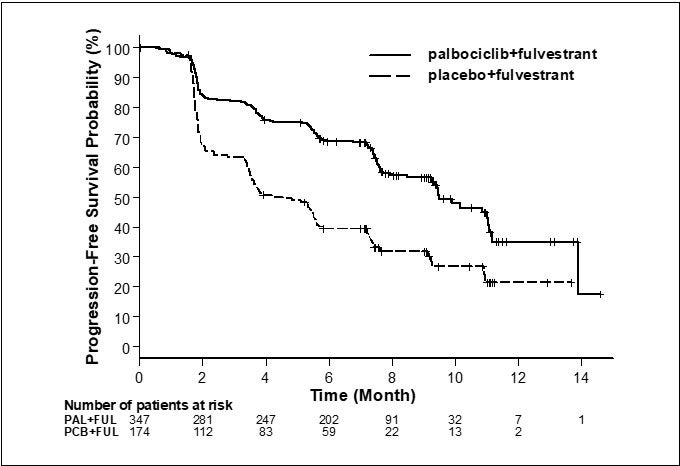
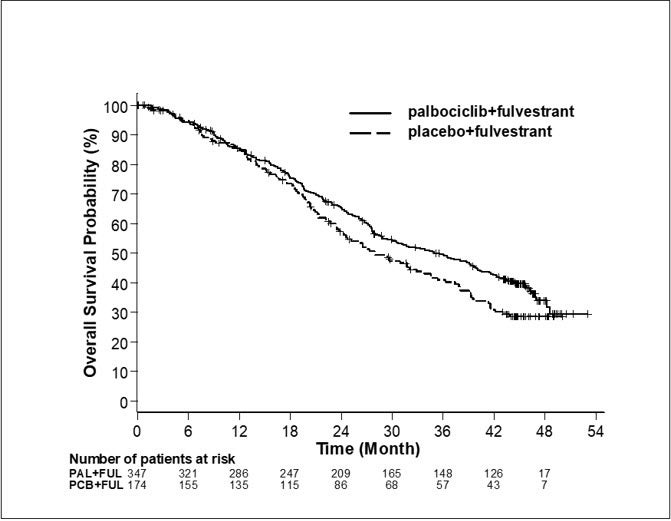
Los resultados de la SLP evaluada por el investigador y la SG final del Estudio 2 se resumen en la Tabla 9. Los gráficos de Kaplan-Meier relevantes se muestran en las figuras 2 y 3, respectivamente. Se observaron resultados consistentes de la SLP entre los subgrupos de pacientes de foco de la enfermedad, sensibilidad al tratamiento hormonal previo y situación respecto a la menopausia. Luego de un tiempo de seguimiento medio de 45 meses, los resultados de la SG final no fueron estadísticamente significativos.
Sobrevida libre de progresión para IT
\
(evaluación del investigador)
Respuesta objetiva para pacientes con enfermedad mensurable
\
(evaluación del investigador)
- *
- Las respuestas se basan en respuestas confirmadas.
- †
- No son significativas estadísticamente en el nivel alfa de 2 lados preespecificado de 0.047.
- ‡
- El valor p de 2 lados de la prueba de rango logarítmico estratificada por la presencia de metástasis visceral y sensibilidad al tratamiento hormonal previo por aleatorización.
16 PRESENTACIÓN/CONSERVACIÓN Y MANIPULACIÓN
IBRANCE se presenta en las concentraciones y configuraciones de envase siguientes:
Conservar entre 20 °C y 25 °C (68 °F a 77 °F); se permiten oscilaciones entre 15 °C y 30 °C (59 °F a 86 °F) [consultar Temperatura ambiente controlada de USP].
17 INFORMACIÓN DE ASESORÍA AL PACIENTE
Advertir al paciente que debe leer el documento para el paciente aprobado por la FDA (Información para el paciente).
Mielodepresión/Infección
- Advertir a los pacientes que deben informar inmediatamente cualquier signo o síntoma de mielodepresión o infección, como fiebre, escalofríos, mareos, falta de aliento, debilidad o mayor tendencia a sangrar o presentar moretones [consultar Advertencias y precauciones (5.1)].
Enfermedad pulmonar intersticial/Neumonitis
- Advertir a los pacientes que deben informar de inmediato los síntomas respiratorios nuevos o si empeoran [consultar Advertencias y precauciones (5.2)].
Interacciones farmacológicas
- La toronja (pomelo) puede interactuar con IBRANCE. Los pacientes no deben consumir productos de toronja (pomelo) mientras estén en tratamiento con IBRANCE.
- Se debe informar a los pacientes que deben evitar los inhibidores potentes del CYP3A y los inductores potentes del CYP3A.
- Advertir a los pacientes que deben informar a sus proveedores de atención médica de todos los medicamentos concomitantes, incluidos los medicamentos de venta con receta, de venta libre, vitaminas y productos a base de hierbas [consultar Interacciones farmacológicas (7)].
Posología y administración
- Advertir a los pacientes que tomen IBRANCE con alimentos.
- Si el paciente vomita o se salta una dosis, no debe tomar una dosis adicional. La siguiente dosis prescrita se deberá tomar a la hora habitual. Las cápsulas de IBRANCE deben tragarse enteras (sin masticarlas, triturarlas ni abrirlas antes de tragarlas). No se deberá ingerir ninguna cápsula que esté rota, agrietada o no esté intacta de alguna otra forma.
- Las mujeres pre/perimenopáusicas tratadas con IBRANCE también deben tratarse con agonistas de la LHRH [consultar Posología y administración (2.1)].
Embarazo, lactancia e infertilidad
- Toxicidad embriofetal
- Advertir a las mujeres con capacidad reproductiva del riesgo posible para el feto y que deben usar anticoncepción eficaz durante el tratamiento con IBRANCE y por al menos 3 semanas después de la última dosis. Advertir a las mujeres que informen a su proveedor de atención médica si quedan embarazadas o si hay sospecha de embarazo [consultar Advertencias y precauciones (5.3) y Uso en poblaciones específicas (8.1 y 8.3)].
- Advertir a los pacientes hombres que tengan parejas mujeres con capacidad reproductiva que deben usar un anticonceptivo eficaz durante el tratamiento con IBRANCE y durante al menos 3 meses después de la última dosis [consultar Uso en poblaciones específicas (8.3)].
- Lactancia: Advertir a las mujeres que no amamanten durante el tratamiento con IBRANCE y por 3 semanas después de la última dosis [consultar Uso en poblaciones específicas (8.2)].
- Infertilidad: Se debe informar a los hombres con capacidad reproductiva que IBRANCE puede causar infertilidad y que consideren preservar el esperma antes de tomar IBRANCE [consultar Uso en poblaciones específicas (8.3)].
El prospecto de este producto puede haberse actualizado. Para obtener la Información de prescripción completa, visite www.Pfizer.com. Para obtener información médica acerca de IBRANCE, visite www.pfizermedinfo.com o llame al 1-800-438-1985.

LAB-0723-9.0
¿Cuál es la información más importante que debo saber sobre IBRANCE?
IBRANCE puede causar efectos secundarios graves, entre ellos:
Recuento bajo de glóbulos blancos (neutropenia). Los recuentos bajos de glóbulos blancos son muy frecuentes cuando se toma IBRANCE y pueden causar infecciones graves que pueden producir la muerte. Su proveedor de atención médica debe verificar sus recuentos de glóbulos blancos antes del tratamiento y durante este.
Si presenta recuentos bajos de glóbulos blancos durante el tratamiento con IBRANCE, su proveedor de atención médica puede interrumpir el tratamiento, disminuir la dosis o indicarle que espere para comenzar el ciclo de tratamiento. Informe de inmediato a su proveedor de atención médica si presenta signos o síntomas de recuentos bajos de glóbulos blancos o de infección, como fiebre o escalofríos.
Problemas pulmonares (neumonitis). IBRANCE puede provocar una inflamación grave o potencialmente mortal en los pulmones durante el tratamiento lo cual puede conducir a la muerte. Informe a su proveedor de atención médica si tiene síntomas nuevos o si empeoraron, entre ellos:
- dolor de pecho
- tos con o sin mucosidad
- dificultad para respirar o falta de aire
Su proveedor de atención médica puede interrumpir o detener por completo el tratamiento con IBRANCE si sus síntomas son intensos.
Consulte “¿Cuáles son los posibles efectos secundarios de IBRANCE?” para obtener más información acerca de los efectos secundarios.
¿Qué es IBRANCE?
IBRANCE es un medicamento de venta con receta que se utiliza en adultos para el tratamiento del cáncer de mama con receptor hormonal positivo (HR+) y receptor 2 del factor de crecimiento epidérmico humano negativo (HER2-) que se ha propagado a otras partes del cuerpo (metastásico) en combinación con:
- un inhibidor de la aromatasa como primer tratamiento a base de hormonas, o
- fulvestrant en personas con progresión de la enfermedad después del tratamiento hormonal.
Se desconoce si IBRANCE es seguro y eficaz en niños.
¿Qué debo decirle a mi proveedor de atención médica antes de tomar IBRANCE?
Antes de tomar IBRANCE, dígale a su proveedor de atención médica todas sus afecciones médicas, incluyendo si:
- tiene fiebre, escalofríos o cualquier otro signo o síntoma de infección.
- tiene problemas hepáticos o renales.
- tiene cualquier otra afección médica.
- está embarazada o tiene previsto quedar embarazada. IBRANCE puede hacerle daño al bebé en gestación.
- Las mujeres con capacidad de quedar embarazadas deben utilizar un método anticonceptivo eficaz durante el tratamiento y, como mínimo, durante 3 semanas después de tomar la última dosis de IBRANCE. Es posible que su proveedor de atención médica le pida someterse a una prueba de embarazo antes de iniciar el tratamiento con IBRANCE.
- Los hombres que tengan parejas del sexo femenino que puedan quedar embarazadas deben utilizar un método anticonceptivo eficaz durante el tratamiento con IBRANCE y, como mínimo, durante 3 meses después de tomar la última dosis de IBRANCE.
- Hable con su proveedor de atención médica sobre los métodos anticonceptivos que puedan ser adecuados para usted durante este tiempo.
- Si queda embarazada o cree que está embarazada, dígaselo a su proveedor de atención médica inmediatamente.
- está amamantando o planea hacerlo. Se desconoce si IBRANCE se excreta en la leche materna. No amamante mientras esté en tratamiento con IBRANCE ni durante 3 semanas después de tomar la última dosis.
Informe a su proveedor de atención médica sobre todos los medicamentos que toma, incluidos los de venta con receta y los de venta libre, las vitaminas y los suplementos a base de hierbas. IBRANCE y otros medicamentos pueden interactuar entre sí y causar efectos secundarios.
Conozca todos los medicamentos que toma. Tenga una lista de dichos medicamentos para mostrársela a su proveedor de atención médica o farmacéutico cuando obtenga un medicamento nuevo.
¿Cómo debo tomar IBRANCE?
- Tome IBRANCE exactamente como se lo indique su proveedor de atención médica.
- Tome IBRANCE con alimentos.
- IBRANCE debe tomarse aproximadamente a la misma hora todos los días.
- Trague las cápsulas de IBRANCE enteras. No mastique, triture ni abra las cápsulas de IBRANCE antes de tragarlas.
- No tome ninguna cápsula de IBRANCE que esté rota, agrietada o que parezca estar dañada.
- Evitar consumir toronja (pomelo) y productos de toronja durante el tratamiento con IBRANCE. La toronja puede aumentar la cantidad de IBRANCE en la sangre.
- No modifique su dosis ni deje de tomar IBRANCE a menos que se lo indique su proveedor de atención médica.
- Si olvida una dosis de IBRANCE o vomita después de tomar una dosis de IBRANCE, no tome otra dosis ese día. Tome la siguiente dosis en su horario habitual.
- Si toma una dosis excesiva de IBRANCE, llame de inmediato a su proveedor de atención médica o diríjase a la sala de emergencias del hospital más cercano.
¿Cuáles son los posibles efectos secundarios de IBRANCE?
IBRANCE puede causar efectos secundarios graves. Consulte “¿Cuál es la información más importante que debo saber sobre IBRANCE?”
Los efectos secundarios más frecuentes de IBRANCE cuando se usa con letrozol o fulvestrant incluyen:
- Los recuentos bajos de glóbulos rojos y los recuentos bajos de plaquetas son frecuentes con IBRANCE. Llame de inmediato a su proveedor de atención médica si presenta cualquiera de estos síntomas durante el tratamiento:
- mareos
- falta de aliento
- debilidad
- sangrado o formación de moretones con más facilidad
- hemorragia nasal
- infecciones (consultar “¿Cuál es la información más importante que debo saber sobre IBRANCE?”)
- fatiga
- náuseas
- llagas bucales
- valores anormales en las pruebas de función hepática
- diarrea
- caída o pérdida del cabello
- vómitos
- erupción cutánea
- falta de apetito
IBRANCE puede causar problemas de fertilidad en los hombres. Esto puede afectar su capacidad de engendrar un hijo. Si esto le preocupa, antes de tomar IBRANCE hable con su proveedor de atención médica sobre sus opciones de planificación familiar.
Informe a su proveedor de atención médica si tiene efectos secundarios que le molestan o no desaparecen.
Estos no son todos los efectos secundarios posibles de IBRANCE.
Llame a su médico para que le asesore sobre los efectos secundarios. Puede reportar los efectos secundarios a la FDA llamando al 1-800-FDA-1088.
¿Cómo debo conservar IBRANCE?
- Conserve IBRANCE entre 68 °F y 77 °F (20 °C a 25 °C).
Mantenga IBRANCE y todos los medicamentos fuera del alcance de los niños.
Información general sobre el uso seguro y eficaz de IBRANCE
A veces los medicamentos se recetan para fines distintos de los descritos en el folleto de información para el paciente. No use IBRANCE para una afección para la que no se recetó. No les dé IBRANCE a otras personas, incluso si tienen los mismos síntomas que usted, porque podría hacerles daño.
Puede pedirle a su farmacéutico o proveedor de atención médica más información sobre IBRANCE redactada para profesionales de la salud.
¿Cuáles son los ingredientes de IBRANCE?
Principio activo: palbociclib
Excipientes: celulosa microcristalina, lactosa monohidrato, glicolato de almidón sódico, dióxido de silicio coloidal, estearato de magnesio y cubierta de gelatina dura de las cápsulas.
Las cubiertas de las cápsulas opacas de color anaranjado claro, anaranjado claro/caramelo y caramelo contienen: gelatina, óxido férrico rojo, óxido férrico amarillo y dióxido de titanio.
La tinta para impresión contiene: goma laca, dióxido de titanio, hidróxido de amonio, propilenglicol y simeticona.
LAB-0724-6.0
Para obtener más información, visite www.Pfizer.com o llame al 1-800-438-1985.
Revisado: Diciembre 2022
Document Id: 54fa32a8-a9aa-4643-a227-ddf95ec4dd05
34391-3
Set id: e0e6412f-50b4-4fd4-9364-62818d121a07
Version: 9
Entrada en efectividad: 20190919
Pfizer Laboratories Div Pfizer Inc